MetaMLST
MetaMLST is a software tool that performs an in-silico Multi Locus Sequence Typing (MLST) analysis on metagenomic samples. MetaMLST achieves cultivation- and assembly- free strain level tracking. MetaMLST is able to detect and trace all the species to which the standard MLST protocol is applicable.
Citation
If you find this tool useful in your research, please cite our paper:
MetaMLST: multi-locus strain-level bacterial typing from metagenomic samples
Nucleic Acids Research 2016 10.1093/nar/gkw837
1 Centre for Integrative Biology, University of Trento, Trento, Italy
2 Computational Biology Unit, Research and Innovation Centre, Fondazione Edmund Mach, San Michele all'Adige 38010, Italy
How it works
MetaMLST works by reconstructing the MLST loci-sequences using the closest reference from the publicly available datsets (PubMLST, MLST.net) and traces the most abundant strain of each species:
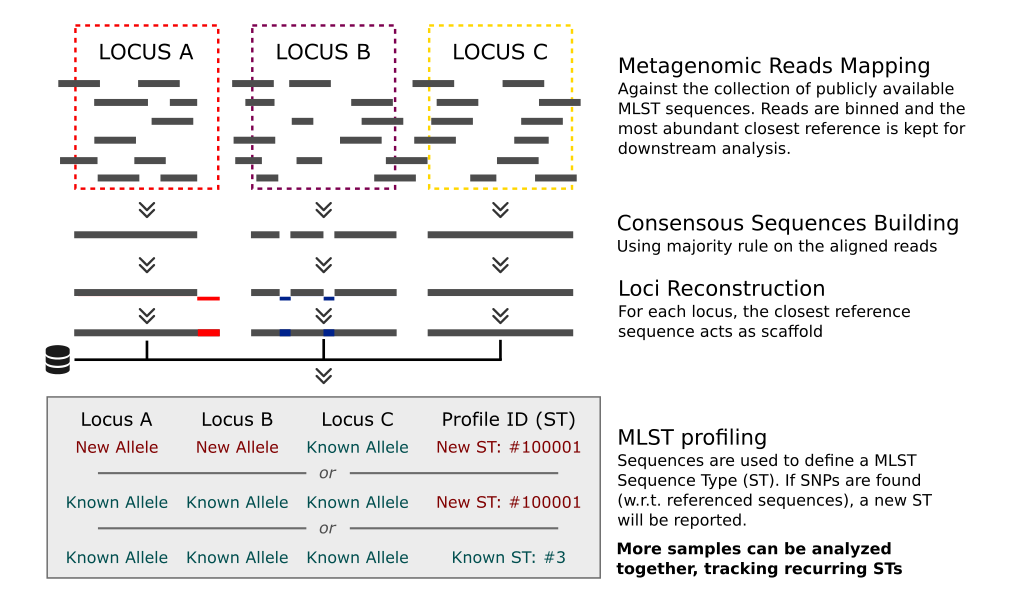
Input / Output
To run MetaMLST on your samples you need:
- NGS Shotgun Sequencing Metagenomic Reads in FASTQ format;
- MLST loci sequences table for your species of interest, in FASTA format, like this;
- MLST profiles table for your species of interest, in TSV format, like this;
- Metadata table for your samples [optional]
MetaMLST produces in output:
- A table with the samples IDs, detected Sequence Types (ST) and the related metadata, if available. (One file for each species detected)
- A tab-separated file containing the updated typing table (i.e. known and newly identified Sequence Types). (One file for each species detected)
- The sequences of the reconstructed MLST loci from the metagenomic source, for each detected type, in many user-selectable formats (separated, concatenated...) for further analyses (e.g. Phylogenetic trees, Minimum Spanning Trees...) like these
Software repository and supporting material
The software repository of MetaMLST is at: https://github.com/SegataLab/metamlst/ For comments and question please refer to the Users Support Group: Here